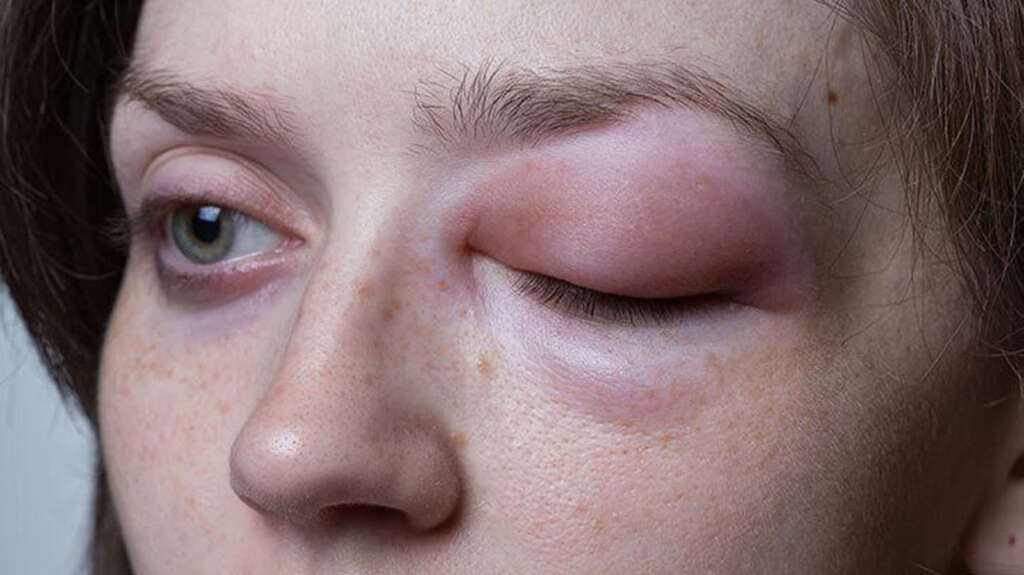
Herediter anjioödem (HAE); nadir görülen, kalıtsal ve potansiyel olarak yaşamı tehdit eden bir hastalıktır. Bu durum, vücutta şişme (ödem) ataklarına neden olan genetik bir bozukluktan kaynaklanır. Genellikle üçüncü basamak komplement sistemi eksikliği veya fonksiyonel bir eksiklik ile karakterizedir.
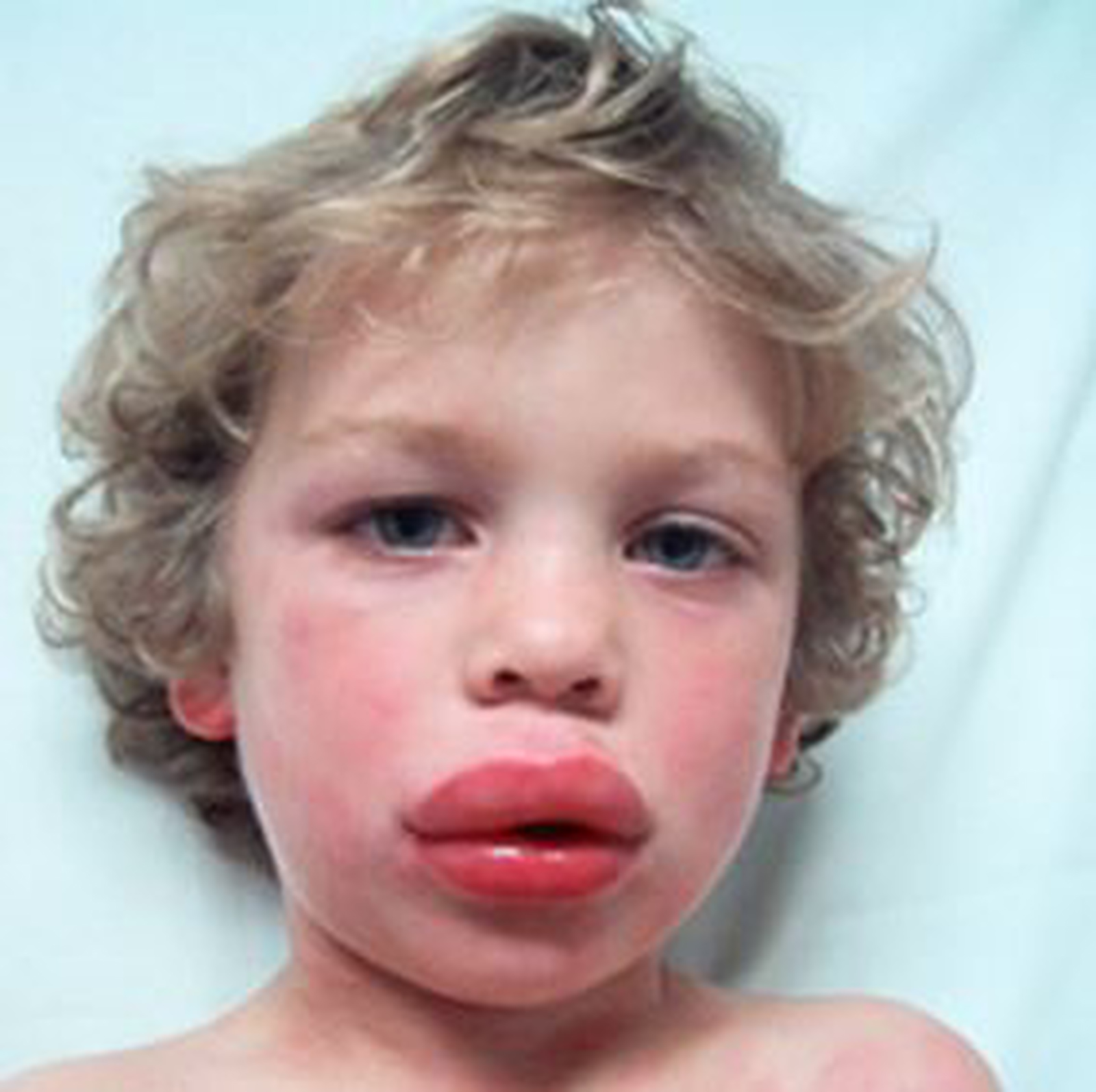
İşte herediter anjioödem hakkında bazı temel bilgiler:
- Belirtiler ve Ataklar: HAE’nin belirgin özelliği, aniden ortaya çıkan ve genellikle yüz, eller, ayaklar, sindirim sistemi veya solunum sistemi gibi vücut bölgelerinde görülen ödem ataklarıdır. Bu ataklar acil tıbbi müdahale gerektirebilir, özellikle boğazda şişme varsa.
- Kalıtım: HAE genellikle genetik bir bozukluk olarak kalıtılır. Hastalık genellikle bir ebeveynden gelen etkilenmiş bir genin çocuğa geçmesiyle ortaya çıkar. Ancak, bazı durumlarda ailesel öykü bulunmaz.
- Genetik Temel: HAE’nin genellikle C1 esteraz inhibitörü adı verilen bir proteinin eksikliği veya işlevsizliği ile ilişkilendirildiği bilinir. Bu protein, vücutta ödem oluşumunu kontrol eden bir sistem olan komplement sisteminin bir parçasıdır.
- Atak Tetikleyicileri: HAE atakları genellikle stres, travma, cerrahi müdahale, hormonal değişiklikler, enfeksiyonlar veya belirli ilaçlar gibi tetikleyici faktörlerle ilişkilidir.
- Tanı ve Tedavi: HAE teşhisi genellikle genetik testlerle konur. Tedavi, akut atakları kontrol altına almaya yönelik ilaçları içerir. Ayrıca, profilaktik tedaviler veya atakları önlemek için kullanılan uzun vadeli ilaçlar da mevcuttur.
- Hastalığın Yönetimi: HAE’nin yönetimi multidisipliner bir yaklaşım gerektirir. Hasta ve sağlık ekibi arasında etkili bir iletişim önemlidir. Hastaların tetikleyici faktörleri tanıması ve kaçınması, durumun yönetiminde önemli bir rol oynar.
Herediter anjioödem, nadir olmakla birlikte ciddi bir durumdur. Hasta bireyler ve aileleri, bu durumu anlamak, yönetmek ve tedavi etmek için bir uzman sağlık ekibiyle işbirliği yapmalıdır. Hastalığın belirtileri olan kişiler, bir sağlık profesyoneli ile görüşmeli ve gerekirse genetik testler yaptırmalıdır.
0
Beğendim
0
Dikkatimi Çekti
0
Doğru Bilgi
0
Eşsiz Bilgi
1
Alkışlıyorum
0
Sevdim




